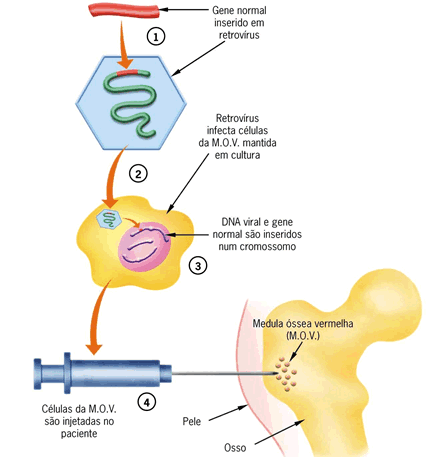
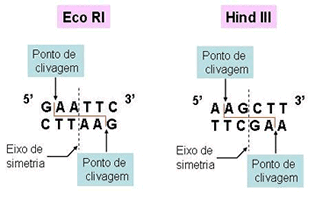
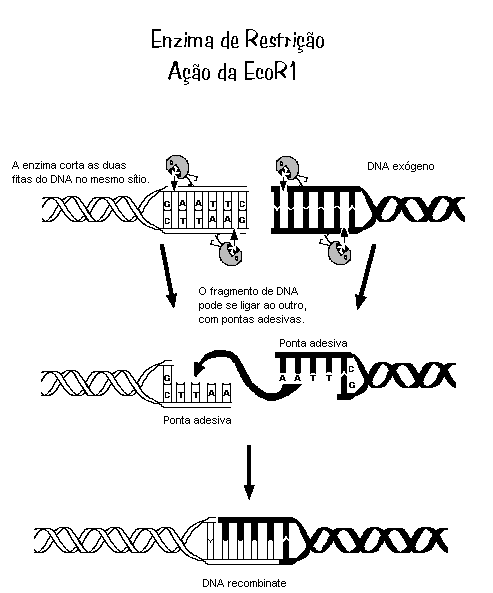
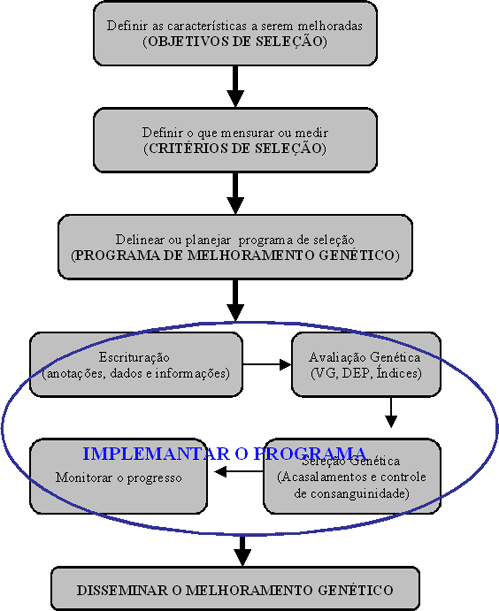
O Mal de Alzheimer, Doença de Alzheimer (DA) ou simplesmente Alzheimer é uma doença degenerativa atualmente incurável mas que possui tratamento. O tratamento permite melhorar a saúde, retardar o declínio cognitivo, tratar os sintomas, controlar as alterações de comportamento e proporcionar conforto e qualidade de vida ao idoso e sua família. Foi descrita, pela primeira vez, em 1906, pelo psiquiatra alemão Alois Alzheimer, de quem herdou o nome. É a principal causa de demência em pessoas com mais de 60 anos no Brasil e em Portugal, sendo cerca de duas vezes mais comum que a demência vascular, sendo que em 15% dos casos ocorrem simultaneamente. Atinge 1% dos idosos entre 65 e 70 anos mas sua prevalência aumenta exponencialmente com os anos sendo de 6% aos 70, 30% aos 80 anos e mais de 60% depois dos 90 anos.
Sintomas
Cada paciente de Alzheimer sofre a doença de forma única, mas existem pontos em comum, por exemplo, o sintoma primário mais comum é a perda de memória. Muitas vezes os primeiros sintomas são confundidos com problemas de idade ou de estresse. Quando a suspeita recai sobre o Mal de Alzheimer, o paciente é submetido a uma série de testes cognitivos e radiológicos. Com o avançar da doença vão aparecendo novos sintomas como confusão mental, irritabilidade e agressividade, alterações de humor, falhas na linguagem, perda de memória a longo prazo e o paciente começa a desligar-se da realidade. Antes de se tornar totalmente aparente o Mal de Alzheimer vai-se desenvolvendo por um período indeterminado de tempo e pode manter-se não diagnosticado e assintomático durante anos.
O Alzheimer é dividido em 4 fases, que são elas:
1º- Os primeiros sintomas são muitas vezes falsamente relacionados com o envelhecimento natural ou com o estresse. Alguns testes neuropsicológicos podem revelar muitas deficiências cognitivas até oito anos antes de se poder diagnosticar o Mal de Alzheimer por inteiro. O sintoma primário mais notável é a perda de memória de curto prazo (dificuldade em lembrar factos aprendidos recentemente); o paciente perde a capacidade de dar atenção a algo, perde a flexibilidade no pensamento e o pensamento abstrato; pode começar a perder a sua memória semântica. Nessa fase pode ainda ser notada apatia, como um sintoma bastante comum. É também notada uma certa desorientação de tempo e espaço. A pessoa não sabe onde está nem em que ano está, em que mês ou que dia. Quanto mais cedo os sintomas forem percebidos mais eficaz é o tratamento e melhor o prognóstico.
 2°(demência inicial) - Com o passar dos anos, conforme os neurônios morrem e a quantidade de neurotransmissores diminuem, aumenta a dificuldade em reconhecer e identificar objectos (agnosia) e na execução de movimentos (apraxia).
2°(demência inicial) - Com o passar dos anos, conforme os neurônios morrem e a quantidade de neurotransmissores diminuem, aumenta a dificuldade em reconhecer e identificar objectos (agnosia) e na execução de movimentos (apraxia).
A memória do paciente não é afetada toda da mesma maneira. As memórias mais antigas, a memória semântica e a memória implícita (memória de como fazer as coisas) não são tão afectadas como a memória a curto prazo. Os problemas de linguagem implicam normalmente a diminuição do vocabulário e a maior dificuldade na fala, que levam a um empobrecimento geral da linguagem. Nessa fase, o paciente ainda consegue comunicar ideias básicas. O paciente pode parecer desleixado ao efetuar certas tarefas motoras simples (escrever, vestir-se, etc.), devido a dificuldades de coordenação.
3°- A degeneração progressiva dificulta a independência. A dificuldade na fala torna-se evidente devido à impossibilidade de se lembrar de vocabulário. Progressivamente, o paciente vai perdendo a capacidade de ler e de escrever e deixa de conseguir fazer as mais simples tarefas diárias. Durante essa fase, os problemas de memória pioram e o paciente pode deixar de reconhecer os seus parentes e conhecidos. A memória de longo prazo vai-se perdendo e alterações de comportamento vão-se agravando. As manifestações mais comuns são a apatia, irritabilidade e instabilidade emocional, chegando ao choro, ataques inesperados de agressividade ou resistência à caridade. Aproximadamente 30% dos pacientes desenvolvem ilusões e outros sintomas relacionados. Incontinência urinária pode aparecer.
4°(terminal)- Durante a última fase do Mal de Alzheimer, o paciente está completamente dependente das pessoas que tomam conta dele. A linguagem está agora reduzida a simples frases ou até a palavras isoladas, acabando, eventualmente, em perda da fala. Apesar da perda da linguagem verbal, os pacientes podem compreender e responder com sinais emocionais. No entanto, a agressividade ainda pode estar presente, e a apatia extrema e o cansaço são resultados bastante comuns. Os pacientes vão acabar por não conseguir desempenhar as tarefas mais simples sem ajuda. A sua massa muscular e a sua mobilidade degeneram-se a tal ponto que o paciente tem de ficar deitado numa cama; perdem a capacidade de comer sozinhos. Por fim, vem a morte, que normalmente não é causada pelo Mal de Alzheimer, mas por outro fator externo (pneumonia, por exemplo).
Prevenção
Todos os estudos de medidas para prevenir ou atrasar os efeitos do Alzheimer são frequentemente infrutíferos. Hoje em dia, não parecem existir provas para acreditar que qualquer medida de prevenção é definitivamente bem sucedida contra o Alzheimer. No entanto, estudos indicam relações entre factores alteráveis como dietas, risco cardiovascular, uso de produtos farmacêuticos ou atividades intelectuais e a probabilidade de desenvolvimento de Alzheimer da população. Mas só mais pesquisa, incluídos testes clínicos, revelarão se, de facto, esses factores podem ajudar a prevenir o Alzheimer.
A inclusão de fruta e vegetais, pão, trigo e outros cereais, azeite, peixe, e vinho tinto, podem reduzir o risco de Alzheimer. Algumas vitaminas como a B12, B3, C ou a B9 foram relacionadas em estudos ao menor risco de Alzheimer, embora outros estudos indiquem que essas não têm nenhum efeito significativo no início ou desenvolvimento da doença e podem ter efeitos secundários. Algumas especiarias como a curcumina e o açafrão mostraram sucesso na prevenção da degeneração cerebral em ratos de laboratório.
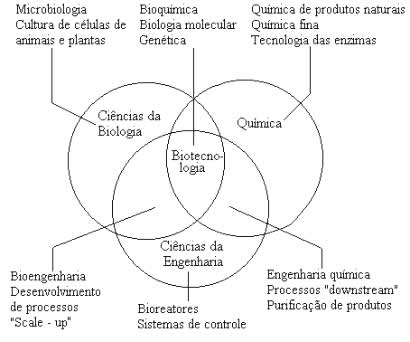
 Posso concluir que ao conhecer essas diversas síndromes e doenças, percebi o quanto é vasto esse mundo e que o ser Humano tem que estudar muito detalhadamente cada assunto desses para entender como acontece, como curar, e como melhorar de certa forma com a medicina a vida dessas pessoas. Com essas postagens também percebemos que o ser Humano não está parado, ele está se desenvolvendo através da biotecnologia, com novas técnicas de manipulação do DNA para melhor se compreender a vida humana e dos outros seres existentes no nosso planeta. Podemos dizer que estamos muito longe, mas o ser Humano está caminhando cada vez mais para compreender a vida e esse vasto mundo de conhecimentos.
Posso concluir que ao conhecer essas diversas síndromes e doenças, percebi o quanto é vasto esse mundo e que o ser Humano tem que estudar muito detalhadamente cada assunto desses para entender como acontece, como curar, e como melhorar de certa forma com a medicina a vida dessas pessoas. Com essas postagens também percebemos que o ser Humano não está parado, ele está se desenvolvendo através da biotecnologia, com novas técnicas de manipulação do DNA para melhor se compreender a vida humana e dos outros seres existentes no nosso planeta. Podemos dizer que estamos muito longe, mas o ser Humano está caminhando cada vez mais para compreender a vida e esse vasto mundo de conhecimentos.