A síndrome Spoan é uma condição de herança autossômica recessiva que até o momento foi reconhecida apenas no Brasil e que caracteriza-se por: 1. paraplegia espástica de início nos primeiros anos de vida e caráter progressivo, 2. atrofia óptica congênita e 3. neuropatia periférica sensitivo-motora axonal, de início a partir da primeira década de vida. Com freqüência ocorrem deformidades esqueléticas, escoliose e sobressaltos à estimulação sonora. A sua caracterização inicial foi feita por nosso grupo que avaliou 25 indivíduos originários do Alto Oeste do Rio Grande do Norte. Estudo de ligação mostrou que o gene responsável pela síndrome Spoan localizava-se em 11q12-13 em uma região com 4,8 Mb. Com o auxílio de agentes do Programa de Saúde da Família, realizamos um estudo epidemiológico em cinco cidades da região (população em 2007: 39.504 habitantes) que estavam listadas entre os 50 municípios com maior prevalência de deficiência no Brasil. Foram colhidas informações a respeito de consangüinidade e deficiência e, após triagem inicial, pacientes com clínica sugestiva de Spoan foram avaliados por médicos especialistas. Em dois municípios (Serrinha dos Pintos e São Miguel) foram identificados 41 pacientes com Spoan. Avaliamos também 27 pacientes com a essa doença, procedentes de municípios que não foram prospectados de forma sistemática. No total, diagnosticamos 68 pacientes com Spoan, frutos de 43 uniões, 40 das quais reconhecidamente consangüíneas. A freqüência de casamentos consanguíneos nos cinco municípios prospectados variou entre 9 e 32,5%. Estudo de ligação com marcadores de tipo microssatélites e SNPs, nos permitiu reduzir as dimensões da região candidata para 2.3 Mb, entre o SNP rs1939212 e o microssatélite D11S987, em 11q13. Para o marcador D11S1889, com alelos em homozigose para todos os pacientes, foi obtido um lod score máximo de 27, para fração de recombinação igual a zero (θ=0.0). Nesta região encontram-se cerca de 70 genes, muitos dos quais com expressão no tecido nervoso. Pretendemos dar continuidade ao refinamento da região candidata e, se possível, identificar o gene responsável pela síndrome Spoan.
domingo, 10 de junho de 2012
Síndrome de Patau
Reconhecida em 1960 por Klaus Patau observando um caso de malformações múltiplas em um neonato, sendo trissômico para o cromossomo 13. Tem como causa a não disjunção dos cromossomos durante a anáfase 1 da mitose, gerando gametas com 24 cromátides. Cerca de 20% dos casos resultam de uma translocação não-balanceada.
A sua incidência foi estimada em cerca de 1 caso para 6000 nascimentos. Aproximadamente 45% dos afetados falecem após 1 mês de vida; 70%, aos 6 meses e somente menos de 5% dos casos sobrevivem mais de 3 anos. A maior sobrevida relatada na literatura foi a de 10 anos de idade.
Assim como a maioria das outras trissomias, associa-se à idade materna avançada, por estarem mais propícias a ocorrência da não disjunção dos cromossomos. A idade da mãe é superior a 35 anos em 40% dos casos.
A trissomia tem origem do óvulo feminino, pelo fato da fêmea maturar geralmente apenas um ovócito, em antagonismo com o macho, que matura milhões de espermatozóides. Gametas masculinos portadores de alterações numéricas cromossômicas tem menor viabilidade que gametas normais, sendo mínimas as possibilidades de um gameta masculino com 24 cromátides fecundar um ovócito.

CARACTERÍSTICA DOS PORTADORES
 O fenótipo inclui malformações graves do sistema nervoso central como arrinencefalia. Um retardamento mental acentuado está presente. Em geral há defeitos cardíacos congênitos e defeitos urigenitais incluindo criptorquidia nos meninos, útero bicornado e ovários hipoplásticos nas meninas gerando inviabilidade, e rins policísticos. Com freqüência encontram-se fendas labial e palato fendido, os punhos cerrados e as plantas arqueadas. A fronte é oblíqua, há hipertelorismo ocular e microftalmia bilateral, podendo chegar a anoftalmia, coloboma da íris, olhos são pequenos extremamente afastados ou ausentes. As orelhas são malformadas e baixamente implantadas. As mãos e pés podem mostrar quinto dedo (polidactilia) sobrepondo-se ao terceiro e quarto, como na trissomia do 18.
O fenótipo inclui malformações graves do sistema nervoso central como arrinencefalia. Um retardamento mental acentuado está presente. Em geral há defeitos cardíacos congênitos e defeitos urigenitais incluindo criptorquidia nos meninos, útero bicornado e ovários hipoplásticos nas meninas gerando inviabilidade, e rins policísticos. Com freqüência encontram-se fendas labial e palato fendido, os punhos cerrados e as plantas arqueadas. A fronte é oblíqua, há hipertelorismo ocular e microftalmia bilateral, podendo chegar a anoftalmia, coloboma da íris, olhos são pequenos extremamente afastados ou ausentes. As orelhas são malformadas e baixamente implantadas. As mãos e pés podem mostrar quinto dedo (polidactilia) sobrepondo-se ao terceiro e quarto, como na trissomia do 18.47, XX (OU XY) + 13
- Deficiência mental
- Surdez; Polidactilia
- Lábio e/ou Palato Fendido
- Anomalias Cardíacas
- Ocorrência 1/10.000
- 88% morre no 1º mês só 5% sobrevive até o 6º mês

Síndrome de Edward
Essa síndrome foi descrita pela primeira vez em 1960 por Edward e colaboradores. Sua prevalência varia entre 1:6.000 a 1:8.000 nascimentos. Cerca de 95% dos embriões portadores da trissomia 18 evoluem para aborto espontâneo ou óbito fetal, nascendo apenas 5%. A mortalidade pós-natal da trissomia 18 é elevada, tendo estes recém-nascidos uma sobrevida média inferior a uma semana; globalmente, menos de 5% destas crianças atingem o primeiro ano de vida.

A causa da trissomia do 18, em cerca de 95% dos casos, é a não separação do cromossomo no momento da formação do gameta – trissomia 18 “livre” – geralmente, o gameta de origem materna. Está relacionada com o aumento da idade materna. São raros os casos em que o cromossoma extra tem origem paterna (erros mitóticos pós fertilização). A origem genética pode ser ainda pode ocorrer por translocação herdada (“de novo”) ou mosaicismo.
O seu diagnóstico pode ser feito ainda intra-útero, no período pré-natal, sendo indicada uma investigação genética no material fetal quando a idade da mãe for superior a 35 anos, alteração nos exames ultrassonográficos (translucência nucal alterada, ausência do osso nasal e outras malformações).
O diagnóstico, após o nascimento, é realizado através do quadro clínico do recém-nascido e do estudo genético.
CARACTERÍSTICAS DOS PACIENTES COM SÍNDROME DE EDWARDS
Fenotípicas (aparência): atraso de crescimento, microcefalia, micrognatia, orelhas dismórficas, onfalocelo, alterações radiais dos membros, dedos caracteristicamente flectidos, proeminência dos calcanhares.
Malformações associadas: cardíacas, cerebrais (quistos do plexo coroideu), osteoarticulares, digestivas (atresia do esófago, divertículo de Meckell), mielomeningocelo.
 CARACTERÍSTICAS
CARACTERÍSTICAS
- Deficiência mental e crescimento
- Hipertonicidade
- Implantação baixa das orelhas
- Mandíbula Recuada
- Rim duplo
- Ocorrência 1/6.000 nascimentos
- 5% a 10% sobrevive o 1º ano
Síndrome de Turner
É uma monossomia na qual os indivíduos afetados exibem sexo feminino mas geralmente não possuem cromatina sexual. O exame de seu cariótipo revela comumente 45 cromossomos, sendo que do par dos cromossomos sexuais há apenas um X; dizemos que esses indivíduos são XO (xis-zero), sendo seu cariótipo representado por 45 X. Muitas dessas concepções terminam em aborto; é provável que 97% desses conceitos sejam eliminados chegando a termo apenas 3%, de modo que essa monossomia constitui uma das causas mais comuns de morte Intra-uterina. Por isso é uma anomalia cromossômica rara, atingindo apenas 1 entre 3000 mulheres normais.

Trata-se, fundamentalmente, de mulheres com disgenesia gonadal, isto é, cujos ovários são atrofiados e desprovidos de folículos; portanto, essas mulheres não procriam, exceto em poucos casos relatados de Turner férteis, em cujos ovários certamente há alguns folículos.
Devido à deficiência de estrógenos elas não desenvolvem as características sexuais secundáriasao atingir a puberdade, sendo, portanto, identificadas facilmente pela falta desses caracteres; assim, por exemplo, elas não menstruam (isto é, têm amenorréia primária). Quando adultas apresentam geralmentebaixa estatura, não mais que 150 cm; infantilismo genital – clitóris pequeno, grandes lábios despigmentados, escassez de pêlos pubianos; pelve andróide, isto é, masculinizada; pele frouxa devido à escassez de tecidos subcutâneos, o que lhe dá aparência senil; unhas estreitas; tórax largo e em forma de barril; alterações cardíacas e ósseas. No recém-nascido frequentemente há edemas nas mãos e nos pés, o que leva a suspeitar da anomalia.
As primeiras observações realizadas com indivíduos severamente afetados associavam a síndrome de Turner algum grau de deficiência mental. Posteriormente ficou evidente que estas pacientes têm um desenvolvimento cognitivo alterado apenas qualitativamente, pois elas possuem uma inteligência verbal superior à das mulheres normais, compensando, assim, as suas deficiências quanto à percepção forma-espaço. Disto resulta que o nível intelectual global das Turner é igual ou, mesmo, levemente superior ao da população feminina normal.
Por outro lado, não exibem desvios de personalidade, o que significa, inclusive, que sua identificação psicossexual não é afetada. Em decorrência da disgenesia ovariana, a única fonte de estrógenos para essas pessoas são as supra-renais; como a taxa desses hormônios é baixa, as pacientes devem receber aplicações de estrógenos para estimular o desenvolvimento dos caracteres sexuais secundários e o aparecimento da menstruação. Usualmente esse tratamento tem início aos 16 anos para evitar que os estrógenos aplicados retardem ainda mais o crescimento.

Síndrome de Klinefelter
São indivíduos do sexo masculino que apresentam cromatina sexual e cariótipo geralmente 47 XXY. Eles constituem um dentre 700 a 800 recém-nascidos do sexo masculino, tratando-se, portanto; de uma das condições intersexuais mais comuns. Outros cariótipos menos comuns são 48 XXYY; 48 XXXY; 49 XXXYYe 49 XXXXY que, respectivamente, exibem 1, 2. e 3 corpúsculos de Barr.
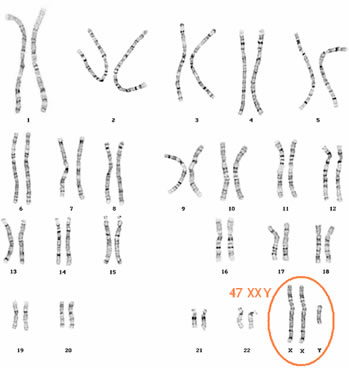
Embora possam ter ereção e ejaculação. são estéreis, pois seus testículos são pequenos e não produzem espermatozóides devido à atro fia dos canais seminíferos. Outras características muitas vezes presentes são: estatura elevada corpo eunucóide, pênis pequeno, pouca pilosidade no púbis e ginecomastia (crescimento das mamas).
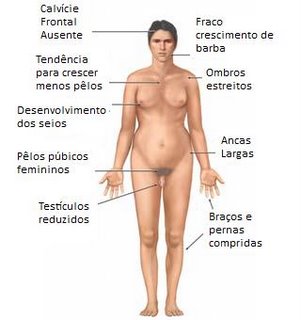 |
Ao contrario do que ocorre na Síndrome de Turner, os pacientes Klinefelter apresentam problemas no desenvolvimento da personalidade, que é imatura e dependente, provavelmente em decorrência de sua inteligência verbal diminuída.
|

Tratamento e Prevenção das Complicações
Este síndrome raramente é diagnosticado no recém-nascido face à ausência de sinais específicos. O diagnóstico precoce permite a intervenção atempada, seja ela psicológica ou farmacológica. O rastreio de problemas visuais, auditivos, assim como a avaliação do desenvolvimento devem ser realizados periodicamente. As anomalias constatadas devem ser seguidas em consultas de especialidade.Muitos destes doentes são referenciados pelos problemas comportamentais, desenvolvimento pubertal anómalo ou infertilidade. A puberdade apresenta problemas particulares secundários aos problemas genitais já referidos. Para uma melhor resposta, o tratamento com testosterona deve ser iniciado pelos 11-12 anos de idade. Está demonstrada a sua eficácia numa percentagem importante de doentes, tanto em aspectos psicossociais como físicos. Este tratamento deve resultar na progressão normal do desenvolvimento físico e sexual, incluindo o crescimento de pêlos púbicos e o aumento do tamanho do pénis e escroto, crescimento da barba, agravamento da voz, aumento do tamanho e da força muscular. Por estes motivos estas crianças e adultos jovens devem ser acompanhados numa consulta de endocrinologia.
Este síndrome raramente é diagnosticado no recém-nascido face à ausência de sinais específicos. O diagnóstico precoce permite a intervenção atempada, seja ela psicológica ou farmacológica. O rastreio de problemas visuais, auditivos, assim como a avaliação do desenvolvimento devem ser realizados periodicamente. As anomalias constatadas devem ser seguidas em consultas de especialidade.Muitos destes doentes são referenciados pelos problemas comportamentais, desenvolvimento pubertal anómalo ou infertilidade. A puberdade apresenta problemas particulares secundários aos problemas genitais já referidos. Para uma melhor resposta, o tratamento com testosterona deve ser iniciado pelos 11-12 anos de idade. Está demonstrada a sua eficácia numa percentagem importante de doentes, tanto em aspectos psicossociais como físicos. Este tratamento deve resultar na progressão normal do desenvolvimento físico e sexual, incluindo o crescimento de pêlos púbicos e o aumento do tamanho do pénis e escroto, crescimento da barba, agravamento da voz, aumento do tamanho e da força muscular. Por estes motivos estas crianças e adultos jovens devem ser acompanhados numa consulta de endocrinologia.
Assinar:
Comentários (Atom)